Simulation of Vibrational Circular Dichroism Spectra Using Second-Order Møller−Plesset Perturbation Theory and Configuration Interaction Doubles
Authors:
Brendan M. Shumberger and T. Daniel Crawford
Affiliation:
Department of Chemistry, Virginia Tech, Blacksburg, Virginia 24061, United States
Description:
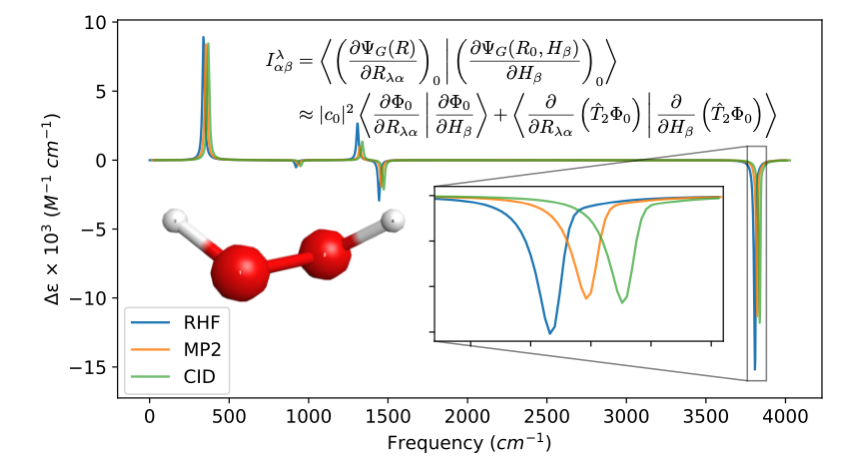
We present the first single-reference calculations of the atomic axial tensors (AATs) with wave function-based methods including dynamic electron correlation effects using second-order Møller−Plesset perturbation theory (MP2) and configuration interaction doubles (CID). Our implementation involves comput- ing the overlap of numerical derivatives of the correlated wave functions with respect to both nuclear displacement coordinates and the external magnetic field. Out test set included three small molecules, including the axially chiral hydrogen molecule dimer and (P)-hydrogen peroxide, and the achiral H2O. For our molecular test set, we observed deviations of the AATs for MP2 and CID from that of the Hartree−Fock (HF) method upward of 49%, varying with the choice of basis set. For (P)-hydrogen peroxide, electron correlation effects on the vibrational circular dichroism (VCD) rotatory strengths and corresponding spectra were particularly significant, with maximum deviations of the rotatory strengths of 62 and 49% for MP2 and CID, respectively, using our largest basis set. The inclusion of dynamic electron correlation to the computation of the AATs can have a significant impact on the resulting rotatory strengths and VCD spectra.
Publications:
- Brendan M. Shumberger and T. Daniel Crawford; Simulation of Vibrational Circular Dichroism Spectra Using Second- Order Møller−Plesset Perturbation Theory and Configuration Interaction Doubles; J. Chem. Theory Comput., 2024
Tags:
Modeling and SimulationRelated Chronicles:
No related chronicles available
Files:
| File Name | File Description | File Type | File Size | File URL |
|---|---|---|---|---|
| Supporting Information | Supporting information for the finite difference approach to dynamically correlated simulations of vibrational circular dichroism. | 227.6 KB | Login to download |